Liver cancer is the second leading cause of cancer-related death worldwide, accounting for approximately 600,000 deaths each year (1). A number of factors including the high infection rates of HBV made China one of the countries with high liver cancer incidence. Hepatocellular carcinoma (HCC) is the most common type of liver cancer. Because of the lack of effective targeted therapeutic methods, the mortality rate of HCC remains high. The five-year overall survival is only approximately 10% (2). Therefore, further elucidating the mechanism of tumorigenesis and development of HCC and identifying novel targets for therapeutic methods to cure HCC is urgent.
DNA double strand breaks (DSBs) is the most detrimental type of DNA damage which can be repaired by two independent and competitive pathways – homologous recombination (HR) and nonhomologous end joining (NHEJ) to maintain the genome integrity. Similar to many other types of cancers, increased genomic instability caused by defects of DNA repair is a potential driver of HCC (3). On the other hand, cancer cells also need to activate DNA repair to cope with the large amounts of DNA damage induced by high replication stress and increased levels of oxidative stress (4, 5). However, due to the lack of mouse models for in vivo analysis of the differences in DSB repair capacity between HCC and adjacent normal tissues, whether DNA repair pathways can be targeted for HCC and other types of cancer treatment has not been assessed.

On October 5, 2020, a paper entitled “Rational combination therapy for hepatocellular carcinoma with PARP1 and DNA-PK inhibitors” was published on The Proceedings of the National Academy of Sciences of the United States of America by Prof. Zhiyong Mao’s group and Prof. Fanglin Sun’s group from the affiliated Shanghai First Maternity & Infant Hospital, and the School of Life Sciences and Technology at Tongji University. In this work, they generated novel knock-in reporter mouse models for measuring in vivo DSB repair efficiency, and demonstrated that both DSB repair pathways were activated in mouse HCCs due to the aberrant upregulation of PARP1 and DNA-PKcs. Further research revealed that targeting the two proteins to block both DSB repair pathways synergistically inhibited HCC growth in both orthotopic HCC mouse and human PDX models, implicating the high potential of the combination therapy in treating HCC.
To analyze the HR efficiency in mouse liver tissues, researchers generated Rosa26HR reporter mice with a GFP-based HR reporter integrated into Rosa26 locus. To study NHEJ in vivo, researchers employed the Rosa26NHEJ reporter mice gifted by Dr. Vera Gorbunova at the University of Rochester (6). With the method of hydrodynamic tail vein injection, they introduced exogenous vectors encoding I-SceI to livers, and expressed I-SceI endonuclease induced DSBs in specific sites of the reporters. Functional GFP can be reconstituted by successful HR or NHEJ. GFP+ cells can be quantified by immunostaining or FACS to calculate the relative HR efficiency. In mouse HCC induced by DEN/CCL4/alcohol, they found that both HR and NHEJ pathways were up-regulated in HCC tissues by 1.6-fold (HR) and 2.5-fold (NHEJ) compared to those in adjacent normal tissues.
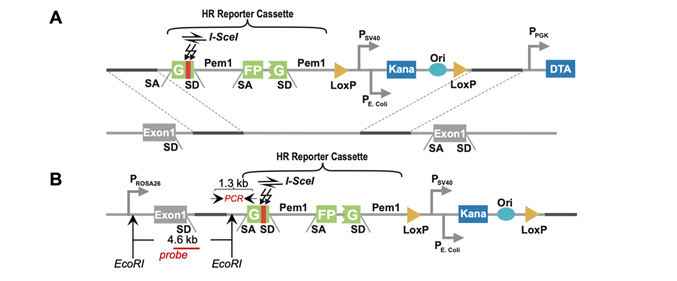
Figure 1 HR reporter for in vivo analysis of HR
In order to elucidate the regulatory mechanisms of up-regulation of HR and NHEJ, researches compared the protein expression of multiple DSB repair factors between mouse HCC and adjacent normal tissues. They found that the expression of PARP1 and DNA-PKcs was up-regulated in mouse HCC. The significant up-regulation of PARP1 and DNA-PKcs was also conserved in human HCC, suggesting that the two factors are particularly critical for activation of DSB repair and the survival of HCC. Further research showed that inhibiting PARP1 with olaparib and DNA-PKcs with NU7441 significantly inhibited the HR and NHEJ pathways in HCC cells. More importantly, inhibiting or depleting both PARP1 and DNA-PKcs synergistically suppressed HCC cell survival in vitro, indicating that they can be potential targets for the treatment of HCC. Further mechanistic studies revealed that inhibiting PARP1 by olaparib suppressed the clearance of nucleosomes at DNA damage sites by blocking the recruitment of ALC1 to DSB sites, thereby inhibiting RPA2 and RAD51 recruitment. Finally, combining olaparib with NU7441 synergistically suppressed HCC growth in both mice and HCC patient-derived-xenograft models (Fig.2).
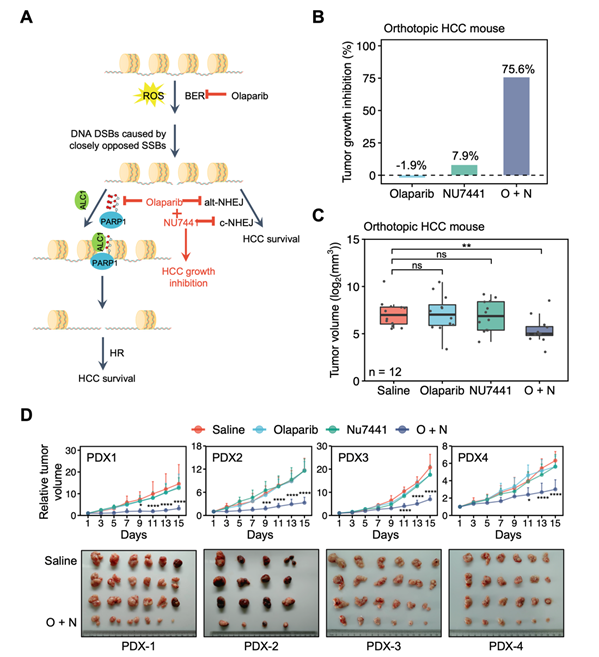
Figure 2 Combination therapy of olaparib and
NU7441 synergistically suppresses the growth of HCC
In summary, they generated novel knock-in reporter mouse models for measuring DSB repair efficiency. The mice would potentially extend our knowledge on DNA repair change and regulatory mechanism of DNA repair in vivo to other types of tumors, as well as in different biological contexts such as aging or other types of diseases. In addition, they also found that PARP1 and DNA-PKcs was critical to the activation of DSB repair in HCC and elucidated the mechanism of PARP1 regulating HR in HCC. Most importantly, the successful preclinical application of combination therapy in HCC indicates that targeting DSB repair holds great potential in treating HCC.
Dr. Chen Wang, Huanyin Tang, Dr. Anke Geng and Dr. Binghua Dai are the co-first authors. Drs. Zhiyong Mao, Fanglin Sun and Ying Jiang are the co-corresponding authors. The work was supported by grants from Chinese National Program on Key Basic Research Project, the National Science Foundation of China, the Fundamental Research Funds for the Central Universities, Program of Shanghai Academic Research Leader “Shuguang Program” of Shanghai Education Development Foundation and Shanghai Municipal Education Commission, Shanghai municipal medical and health discipline construction projects etc.
Source: https://www.pnas.org/content/early/2020/10/02/2002917117
1. L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet‐Tieulent, A. Jemal, Global cancer statistics, 2012. Ca Cancer J Clin. 65, 87–108 (2015).
2. S. F. Altekruse, K. A. McGlynn, M. E. Reichman, Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States From 1975 to 2005. J Clin Oncol. 27, 1485–1491 (2009).
3. N. C. Teoh, Y. Y. Dan, K. Swisshelm, S. Lehman, J. H. Wright, J. Haque, Y. Gu, N. Fausto, Defective DNA strand break repair causes chromosomal instability and accelerates liver carcinogenesis in mice. Hepatology. 47, 2078–2088 (2008).
4. G.-Y. Liou, P. Storz, Reactive oxygen species in cancer. Free Radical Res. 44, 479–496 (2010).
5. H. Gaillard, T. García-Muse, A. Aguilera, Replication stress and cancer. Nat Rev Cancer. 15, 276–289 (2015).
6. A. Vaidya, Z. Mao, X. Tian, B. Spencer, A. Seluanov, V. Gorbunova, Knock-In Reporter Mice Demonstrate that DNA Repair by Non-homologous End Joining Declines with Age. Plos Genet. 10, e1004511 (2014).